
Everything in biology is fluid. At the molecular level, we are constantly moving, flexing and churning inside. But we often don’t think about how proteins change shape—or, as Professor Dorothee Kern, PhD, puts it, how proteins “dance.” Kern, who is also a Howard Hughes Medical Investigator and former professional basketball player, joined the Scripps Research faculty earlier this year from Brandeis University. Her overarching goal is to understand how this dancing influences our health.
“I eat and breathe proteins,” she jokes.
Kern recently published two papers in the Proceedings of the National Academy of Sciences (PNAS), both of which enrich our understanding of the protein field. She sat down with Scripps Research Magazine to explain more about these publications, her upcoming projects here at the institute, and just how important understanding a protein’s choreography is.
What does it mean for a protein to dance?
Throughout my career, what I’ve really tried to promote is the idea that proteins don’t sit in one unique structure. They sample the energy landscape, meaning they’re always hopping from one conformation to the other. This is actually the key to their function: both the dancing between their different structures, as well as how fast this dancing occurs, as it controls the speed of the biological functions. I call it the choreography of a protein’s dance because it’s not random. These movements are highly correlated.
As we showed in our Nature paper published late last year, we developed an algorithm to predict a protein’s different conformations. We have shown over and over that these conformational substates are essential for biological function. That means that learning how proteins “dance” is key to understanding the difference between health and disease.
You had two papers publish recently in PNAS—the first of which delves into protein language models. Can you share what you found?
In this first PNAS paper, we wanted to figure out why certain protein language models work. The models we investigated are fed a single protein sequence and then predict what the corresponding 3-D structure looks like. Yet, how they arrive at these conclusions was a “black box” of sorts.
In this paper, we were determined to figure out how these models learn and predict, if we are to use them reliably as a field. We had three hypotheses: the first, that the model has learned the physics of protein folding and how the atoms are interacting. This was very unlikely, and we could rule it out very quickly. The second hypothesis was that the model actually looks up the entire folds for each protein family. The third one—which turned out to be true—is that it learned to find paired interacting protein fragments, including whole segments.
Big picture, we shed light into the question of how these protein language models learn. On the more technical side, we determined how long the protein segments must be for the model to identify the correct 3-D structure. Machine learning language models, including the Nobel Prize-winning AlphaFold, are so hot right now because solving protein structures with a single sequence has been a monumental breakthrough. But throughout my entire career I never have wanted to blindly use methods without actually knowing the physics behind how they work. I want to call out that this work was led by my postdoc Hannah Wayment-Steele, PhD, together with a visiting student Zhidian Zhang and in a great collaboration with Sergey Ovchinnikov, PhD, assistant professor at MIT.

How does the second PNAS paper build on your previous Nature study?
Protein language models like AlphaFold are limited because they only come up with one structure. The “signal” for the other protein structure—the one it “dances” between—gets diluted. Because I have been working on evolution for a long time now, I said, “Why don’t we get the evolutionary signals for both structures, and how can we accomplish that?”
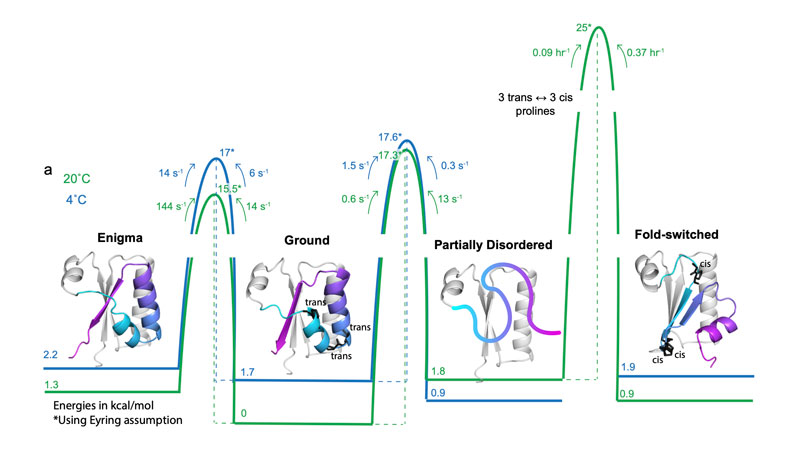
As outlined in the latest Nature study, we developed that novel methodology—which we call “AF-cluster”—using the protein KaiB as a benchmark. KaiB is essential for regulating circadian rhythm in certain bacteria, which I find very fascinating because the atmospheric 24-hour clock is one of the biggest biological drivers. KaiB only has two protein conformations, and if you put it in a test tube with its partner KaiC, they create a 24-hour oscillation—the underlying mechanism we revealed in another Nature paper in early 2023. In this Nature paper, however, we had only predicted KaiB’s two end states—not the actual dynamics, or pathway, of how it went from one conformation to the other.
In our new PNAS study, we looked at exactly how KaiB “travels over the mountain,” so to speak. In other words, how it climbs over the free energy landscape.
What did KaiB’s mechanism reveal?
First of all, we figured out that KaiB’s conversion to its alternate state takes hours. Now here’s another wrong perception in evolution: Many people think that faster is better, but it’s not. The speed of any protein conformational transition is tuned to the biological function needed. The KaiB protein controls the organism’s 24-hour clock, meaning it needs to move super, super slow. In our paper, we saw what evolution had to do to slow this process down.
We studied these changes using nuclear magnetic resonance (NMR)—an amazing method where you can measure protein dynamics in solution at atomic resolution—and we found that KaiB’s conversion takes three hours to be completed. And then we determined the atomistic pathway, which was very complicated. At the high level, we figured out how evolution tunes protein kinetics to align with its overall function.
Credit: Dorothee Kern, Scripps Research
Why is understanding protein dynamics so critical?
I’m always fascinated about how proteins can go between conformations because these changes dictate biology. The rate of how fast the protein works dictates how fast the biological process goes. This means that if you ever want to design enzymes or other proteins—which actually have to flip between states to exert a biological function—we have to understand the process and how the timescale is tuned.
How are you building on this work here at Scripps Research?
First of all, I’m really excited to be in this environment now because of all of the opportunities to collaborate across the institute. The next big thing we’re focused on is predicting protein dynamics using AI. The reason why this is so hard is because there’s not enough data yet to train models. AlphaFold and other language models work well because there are so many protein sequences and structures to feed them. What’s super interesting to me is how to create enough new data to build these predictive models for protein dynamics.
We then want to use this knowledge to work with scientists like Professor Ben Cravatt, PhD. Once we’re able to predict how proteins move, we can move towards drugging different targets involved in various diseases, as his lab is working on.
The other big aspect is using this knowledge to design enzymes. We’re doing this in both ways: rational design and directed evolution. We’re already working with Assistant Professor Ahmed Badran, PhD, and President and CEO Peter Schultz, PhD, to scale up our directed evolution approach, as well as new tactics for increasing the power of biocatalysts for biotechnology applications. It’s been amazing working with people across labs, where we have similar interests in different systems.
You’ve founded multiple oncology companies, including Relay Therapeutics and MOMA Therapeutics. In addition to cancer, are there other diseases you’re focused on?
I founded Relay Therapeutics based on a biophysics concept. The big vision is using differential protein dynamics to make selective drugs, which means you can use the platform for any disease.
One big area outside of cancer we’re focused on is metabolic disease, and we also want to go into brain diseases. For those, you often you have to restore a function, meaning you need therapies that activate their targets (in cancer, most of the time you try to suppress). We published two proof-of-concept papers: In the first one we show how to design activators using allosteric networks—systems where binding at one site of the protein affects activity at a different site. In the second one, we demonstrated that by combining allosteric inhibitors with traditional active site inhibitors, one can combat one of the most devastating problems in cancer treatment: the development of drug resistance.
I like these hard problems where people say it can’t be done. It motivates me!
And finally, what’s your favorite protein?
Adenylate kinase, which is an essential enzyme for every organism and cell as it helps regulate cellular energy. I chose this enzyme when I started as an assistant professor at Brandeis. I gave myself a few months to really think very deeply about which protein to choose to develop all the technologies we needed to describe the full energy landscape—to understand conformational substates and real-time protein dynamics. It’s the only protein ever where all of these substates and analyses are known.